1浙江中医药大学研究生院 浙江 杭州 310016
2 绍兴文理学院附属医院 放射科 312000
Neurofibromatosis typeⅠwith undifferentiated pleomorphic sarcoma of spleen:a report of one case
关键词 Ⅰ型神经纤维瘤病 脾脏 未分化多形性肉瘤
患者 女性,48岁,16年前确诊Ⅰ型神经纤维瘤病,本次因“发热一周,发现脾脏占位3天”入院。查体:T:38.9℃,P:131次/分,BP:117/72mmHg。颜面部及全身皮下可见大小不等的包块,质中等,活动度可。腹软,左侧肋下可触及肿大的脾脏,无压痛。实验室检查提示:血常规:WBC 10×10^9/L,RBC 2.97 ×10^12/L,HGB 64g/L,PLT 493×10^9/L;超敏C反应蛋白310.2mg/L;血沉120mm/H;肿瘤标志物:铁蛋白>2000ng/ml,神经元特异性烯醇化酶48.92ng/ml,其余肿瘤标志物CA125、CA199、CEA、AFP等均在正常范围内;生化检查等未见明显异常。超声检查示:脾脏内探及一大小约108mm×80mm囊实性肿块,边界清,形态欠规则。全腹增强CT示:脾脏肿大,脾内可见巨大囊实性肿物,与正常脾脏结构分界不清,内可见多发分隔,增强后实性部分轻度-中度强化,渐进性强化;脾门处、腹膜后未见明显肿大的淋巴结(图1A、1B、1C)。增强MRI示:脾脏内见巨大肿物,病灶内见大片坏死区,实性部分主要位于病变外周,以T1WI稍低信号,T2WI稍高信号为主,内夹杂线样、小片状T1WI高信号,T2WI低信号影,增强后可见病灶实性部分持续性中度强化,冠状位显示病灶局部突破膈肌(图1D、1E、1F、1G)。CT与MRI均考虑脾脏恶性肿瘤。
患者行脾脏切除及膈肌修补术,术中见:脾脏肿大,大小约20cm×15cm×8cm,脾脏上极见一肿块浸润膈肌。术后免疫组化示:肿块:SMA(-),Vim(+),CD34(血管+),Desmin(-),Melan-A(-),CD68(+),Actin(-),CD31(+),EMA(-),S-100(-),CAM5.2(-),Ki-67(+30%),CD21(-),CD23(-),LCA(散在+),HMB45(Melanoma)(-),CK(Pan)(-)。囊腔:D2-40(-),ERG(血管+),F8(血管+)。病理结果提示:脾脏恶性肿瘤,考虑未分化多形性肉瘤伴坏死(图1H)。
患者术后2月复查CT时发现左肝及左中上腹腔内多发转移瘤,术后7月头颅MRI检查示小脑转移瘤,术后9月死亡。
讨论:神经纤维瘤病(neurofibromatosis,NF)是一种常染色体显性遗传疾病,是由NF1肿瘤抑癌基因突变所致。NF1基因及其蛋白具有抑制肿瘤的作用,NF1基因突变后,失去正常功能的神经纤维素蛋白通过相应的肿瘤通路导致细胞增殖,而细胞凋亡受抑制,最终促进肿瘤的生成[1]。因此,神经纤维瘤病患者罹患肿瘤风险较常人高,但一般以并发中枢神经系统肿瘤、胃肠道间质瘤、乳腺癌、白血病等多见,本例合并发生脾脏未分化多形性肉瘤实属罕见。
未分化多形性肉瘤(undifferentiated pleomorphic sarcoma,UPS)以往被称为恶性纤维组织细胞瘤(milignant fibrous bistiotoma MFH),是起源于间叶组织的恶性肿瘤。好发于四肢及腹膜后,原发于脾脏者罕见。本病以中老年人多见,男性略多于女性[2]。临床表现主要为脾脏肿大和脾脏破裂,也可表现为左上腹不适、疼痛、上腹部肿块、发热、消瘦、腹水等。据文献报道[3],UPS发生于四肢表浅部位、且无复发和转移者治疗效果好,手术切除后生存率高,而发生于内脏及深层组织者恶性程度高,预后差。本例患者病变位于脾脏,发现时病变即已侵犯膈肌,术后一年内出现多个脏器的远处转移及死亡,预后不良,与文献报道相符。
脾脏UPS的CT和MRI图像缺乏特异性,主要表现为脾脏肿大,脾脏内肿块影,直径一般>5cm,形态不规则,边界不清,密度多不均匀,常见坏死、出血、囊变,但钙化少见,其中坏死几乎见于所有肿瘤[4],以病灶中央坏死多见,原因可能为UPS肿瘤细胞增殖活跃,恶性程度高,生长速度快,病变内血供相对不足。增强扫描肿块内实性部分轻-中度强化,渐进性强化,强化特点可能与病变内含有胶原纤维有关。
由于脾脏结构较特殊,脾脏内恶性肿瘤较少见,脾脏UPS需与脾脏淋巴瘤、血管肉瘤等鉴别。1、脾脏淋巴瘤:是脾脏最常见的恶性肿瘤,包括脾脏原发性淋巴瘤和全身性淋巴瘤脾脏浸润,后者较多见。临床表现为脾大、左上腹痛、 发热、体表淋巴结肿大等。实验室检查可以出现白细胞和血小板减少等。脾淋巴瘤可分为均匀弥漫型、粟粒结节型、多发肿块型、单发肿块型
[5],脾脏UPS主要与单发肿块型鉴别,单发肿块型脾淋巴瘤影像表现为:左上腹巨大占位,边界不清,偶见有小片状坏死,出血及钙化罕见,增强均匀或不均匀强化,合并脾外病变时对诊断有很大帮助。脾脏UPS在临床表现及实验室检查上与脾脏淋巴瘤难以区分,影像表现上脾脏UPS坏死常见,极少伴有腹膜后淋巴结肿大,与典型的淋巴瘤可以鉴别。2、脾脏血管肉瘤:好发于中老年人。临床表现为脾脏迅速增大伴左上腹痛,贫血、肝脏肿大及体重减轻。影像表现为:脾脏明显肿大,密度或信号不均,可见单发或多发肿块,边界不清,可见坏死和出血,散在针尖样钙化,也可为大片钙化呈放射状,增强扫描肿瘤呈边缘强化,实性部分不均匀强化,可有囊变坏死,也可自病灶边缘向中心扩展式强化。脾脏血管肉瘤易破裂,引起脾包膜下积血或腹腔积血。易出现临近脏器的侵犯及远处转移。脾脏UPS与之鉴别困难,确诊有赖于病理及免疫组化。
脾脏UPS发病率低,影像表现缺乏特异性,术前误诊率高,影像科医生在影像诊断时仔细辨别病变与膈肌及邻近脏器的关系,仔细观察有无脉管侵犯,有助于临床为患者提供个性化治疗提供依据。
[参考文献]
[1]Khelifa I,Saurat JH,Prins C.Use of imatinib in a patient with cutaneous vasculopathy in the context of von Recklinghausen isease/neurofibromationsis[J].Br J Dermatol,2015,172(1):253-256.
[2]陈涛,严静东,雷贞妮. 未分化多形性肉瘤的影像诊断与鉴别51例[J].实用医学杂志.2016.32(5):789-792.
[3]周雪峰,王雅杰,路红社.恶性纤维组织细胞瘤24例临床分析[J].第二军医大学学报,2001,22(5):474-474.
[4]李娴,孙新海,朱来敏,等.多形性未分化肉瘤的MR表现[J].医学影像学杂志,2014,24(8):1354-1357.
[5]徐秀芳,余日胜.脾脏淋巴瘤的CT诊断.实用放射学杂志[J].2006,22(5):546-548.





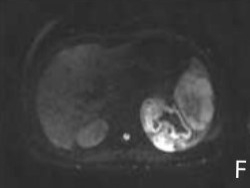


图1 脾脏未分化多形性肉瘤CT、MRI及病理学表现
A、B、C分别为CT平扫、动脉期、静脉期横断位图像,可见脾脏肿大,脾内巨大低密度肿块,边界不清,可见大片坏死区,增强后实性部分轻度-中度强化,渐进性强化;D、E、F、G分别为快速自旋回波T1WI、T2WI、DWI及增强扫描冠状位图像,可见病灶内条索状短T1、长T2信号,提示病灶内出血,冠状位可见病灶突破膈肌;H为病理图(H-E染色,×40),肿瘤细胞排列紧密。

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



