海南双成药业股份有限公司 海南海口市 570314
【摘要】我国药品注册与管理的相关机制与流程较为严格,而严谨的管理为确保药品的安全性与有效性提供重要的制度保障,同时也是对药品研发成果的重要检验。随着药品注册管理新政的不断推进,我国药品研发申报的开展也不可避免地受到了影响。因此,为了有效地进行药品研发申报,需要对药品注册管理新政进行深入地了解与研究,有助于提高药品研发申报的成功率及效率,进而提升企业的经济效益。
【关键词】药品注册;管理新政;药品研发;影响
前言
药品的研发并非一蹴而就,其间必定会经过许多研究和探索,存在很多不确定的风险,如果能够在药品研发过程中有效地进行风险控制,不仅能帮助企业节省人力、财力以及物力等资源的投入,同时有助于提升药品研发申报的成功率及效率,进而提升企业的经济效益。若药品注册申请人能对药品注册管理新政进行深入、透彻地分析与研究,并与国家药品监督管理局药品审评中心(简称CDE)进行积极、有效地沟通,可在一定程度上有效地规避药品研发中存在的风险[1],提高药品研发申报的成功率。随着药品注册管理新政的不断推进,我国药品监督管理相关部门制定并颁布了一系列有关药品研发的技术指导原则、注册申报法规、相关评审程序以及沟通机制等具有改革性的文件,以便更好的指导企业开展药品研发申报,及有效地促进申请人同CDE之间的沟通,呈现出药品注册管理新政对药品研发申报的重要性。
1. 我国药品注册管理新政的推进情况
药品注册是指药品注册申请人依照法定程序和相关要求,提出药物临床试验、药品上市许可、再注册等申请以及补充申请,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,决定是否同意其申请的申报活动[2]。为了更好地开展我国药品注册管理工作,提高审评审批质量、解决注册申请积压、提高仿制药质量、鼓励企业研究和创制新药、健全审评质量控制体系,进一步满足人们对相关药品的需求,更好地助力药品品质的提升[3]。国务院于2015年8月19日颁布实施了《关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号),在这一文件的指导下,国家药品监督管理局(简称NMPA)随之修订并颁布了如《中华人民共和国药品管理法》以及《药品注册管理办法》等相关法律法规管理条例。
至2020年,最新版《药品注册管理办法》已正式发布及落地实施,与之前的管理机制相比,管理新政中,更侧重于对药品注册申报工作的管理,在完善审评审批体系的前提下,对药品注册检验等环节与流程作出进一步的明确。此外,管理新政中,还对药品研发审批程序进行了简化(如原料与制剂关联审评审批),同时落实申请人的主体责任,以及对审评审批过程中,涉及到的技术问题有较明确的相关指导原则指导。这一系列变革,在很大程度上增强了相关管理机制的灵活性,进而有效的保障新政能够有效落地实施。
在管理新政的推行下,2022年我国的医药行业领域处于不断地升级与转型阶段,向更高质量的发展不断迈进,全国范围内的上市公司与龙头企业的研发资金投入高达1202亿元,而且1类创新药品的临床申报数量达到了944种品种,上市申报的种类达到40种,获批的达到21种。
2. 药品注册管理新政在药品研发申报中的作用
2.1对新药及仿制药进行界定,对药品研发热点形成导向
为鼓励新药创制,严格审评审批,提高药品质量,促进产业升级,2016年3月9日总局发布《关于化学药品注册分类改革工作方案的公告》(2016年第51号),对化学药品注册分类进行调整,该文件明确规定了新药和仿制药的申报类别。为落实《化学药品注册分类改革工作方案》要求,规范申请人按照化学药品新注册分类做好注册申报工作,2016年5月4日发布《关于化学药品新注册分类申报资料要求(试行)的通告》(2016年 第80号)以及后续发布的如《M4:人用药物注册申请通用技术文档(CTD)》的通告,均对新药和仿制药的申报研究工作进行了明确的要求和界定。制药企业在对仿制药的研发时,不仅需要根据产品特性进行研究,同时需要对共性部分进行研究,特别是对仿制药与原研药的质量和疗效进行对比研究,确保仿制药与原研药的一致性。而新药研发则侧重于创新和临床研究,一般在研发过程中需经过系统的药理毒理实验,对药品的有效性进行严格地验证,才能有效保障该药物的可开发性。在进行新药临床实验研究前,药物研发者需对药学方面进行研究,不断完善药学研究的相关资料,例如,提供为期12个月的稳定性数据,在评审过程中需继续补充后续的稳定性资料以及补充其他相关研究资料,经CDE审评审批通过并获得批准临床批件后,方可开展临床试验研究以及后续的上市申报研究[4]。此外,管理新政还对评审时间适当地作出调整,药物临床试验申请、药物临床试验期间补充申请的审评审批时限为六十日,药品上市许可申请审评时限为二百日,其中优先审评审批程序的审评时限为一百三十日。总之,药品注册管理新政对仿制药和新药的管理和要求各不同,侧重点也不同,药品注册申报人需了解相关的要求进行药品研究申报。
2.2 技术指导作用
在注册管理新政的指导下,相关部门还颁布了一系列较为明确的技术指导原则与要求,对相关药品的注册审批和研发及生产加大管理与指导力度。例如发布的《化学合成多肽药物药学研究技术指导原则(试行)》的通告(2023年第12号)中便对原料及制剂处方工艺和质量研究控制等进行了规定与指导,在《化学仿制药注册批生产规模的一般性要求(试行)》中对口服制剂、注射剂等药品的注册批与拟定商业化生产规模作出明确的规定。相关规定的发布与实施为药品研发申报提供可参考的技术标准和指导意见,进一步加强了药品注册管理及药品研究技术的规范性,促进药品研发和生产水平的提升,提高药品注册及审评审批技术要求,推动药品注册技术标准国际接轨。
2.3沟通与协调作用
管理新政作为纲领性的指导文件,对制药企业药品研发申报工作给出了一定的指导方向,如2020年CDE颁布关于《药物研发与技术审评沟通交流管理办法》的通告(2020年第48号),对申请人与CDE的沟通交流形式进行了明确。而双方是否需要进行沟通交流,主要是基于药物研发中,某特定阶段关键性技术问题、重大的决策或重要的科学问题等,保障申请人能够获得有关研究中止、暂停或推进的有关建议和意见,这方面的沟通主要以会议的形式开展。除了会议形式的沟通外,对一般的技术问题或是需要补充有关资料等方面问题,申请人还可以通过电话、邮箱或“申请人之窗”进行沟通,提供了多种多样的沟通交流形式。除此之外,CDE还开展相关的培训或咨询会议活动,CDE官网也定期公布各项通知、政策解读、指导原则、意见征求信息以及常见性问题的回复等,申请人可及时下载学习,这也是CDE和申请人进行交流与沟通的方式之一,可将之称为“查询式”沟通。企业需不断深入挖掘其中的细节,根据新政要求完善药品注册研究申报相关工作,提高药品研发申报的成功率,为企业带来良好的经济效益。
3.药品注册管理新政对药品研发申报的影响
3.1 促进药品研发资源向创新药品发展
医药行业作为当代以创新发展内驱力的技术型行业,随着人民群众对身体健康的不断重视及对药品药物需求的多样化,对创新类药物的需求量不断扩大。目前,我国的药品质量已经同国际通用技术标准接轨,应进行实质上的激励措施,以鼓励医药研发创新,不断有效地提升我国创新类药品研发的整体水平。根据CDE每年发布的年度药品审评报告数据统计分析可知,药品注册管理新政颁布与实施以来,我国创新类药品的研发态势喜人,自2017年起,我国新药品注册申请量不断上升。根据《2021年度药品审评报告》(下称报告)中相关数据显示,2021年受理创新药注册申请1886件(998个品种),同比增长76.10%。以药品类型统计,化学药仍为主体,创新化学药1166件(508个品种),同比增长55.05%;创新中药54件(51个品种),同比增长134.78%;创新生物制品666件(439个品种),同比增长125.00%。以注册申请类别统计,新药临床试验申请(以下简称IND)1821件(953个品种),同比增长79.23%;新药上市许可申请(以下简称NDA) 65件(45个品种),同比增长18.18%。《报告》显示,创新药审评数量、药品注册申请按时限审结率均创新高,说明药品注册管理新政对创新药的研究与申报起到促进作用[5]。随着临床研究的发展,创新药品的需求量随之增加,我国药品研发企业应充分参照管理新政,规划更加科学合理的发展途径,有效助推我国的医药行业健康发展,提升其国际竞争能力。
3.2 仿制药研究申报应注重质量与疗效一致性和临床需求
药品注册管理新政的落实,使得仿制药的申报流程得以优化与完善,同时仿制药申报中,所涉及的药品种类及功效也越发注重对临床需求的参考。此外,管理新政还着重强调了仿制药质量和疗效需与原研药一致,不得因仿制而造成药物质量的下降。为了落实仿制药质量和疗效与原研药的一致性,加强一致性评价工作的指导,CDE颁布了一系列政策法规,还专门开通仿制药质量与疗效一致性评价专栏。另外,还多次进行相关技术标准和要求的培训等活动,这足以说明药品注册管理新政对仿制药质量和疗效与原研药一致性的重视程度。注册管理新政不仅对仿制药注册申报进行管理,还对过去已获批上市的产品进行规定和管控,企业需对已上市产品进行一致性评价研究申报,以证明产品质量的安全性和有效性,同时,企业可以结合市场需求进行评估该产品是否有必要开展一致性评价研究。因此,医药企业需对管理新政的具体落实情况进行充分、科学地分析,立足于实际,保障我国医药行业得以有序、健康以及可持续发展。此外,企业在提升自身创新能力水平的同时,应注意仿制药研究申报及根据市场需求进行适度的转型,即向临床需求的方向靠拢,同时需不断提高仿制药质量与疗效。
3.3影响我国药品注册申请量
一般而言,药品注册申请数量能够对当时制药行业药品研发相关活动的整体效率及活跃度等进行一定的反映,随着我国“十二五”规划的发展,CDE受理的相关药品注册申请数量就得到了不断地上升,平均每年都会达到超过7000件的数量,并且屡创新高,例如在2021年受理需技术审评的注册申请9235件,同比增长29.11%。2017至2021年间的注册申请受理量变化图1所示
[5]。
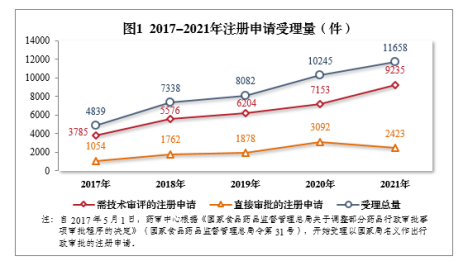
图1 2017-2021年注册申请受理量
随着药品注册管理新政的推进,NMPA对审评审批程序不断完善, 2019至2021年期间的注册申请量有明显的增长,因此可以直观地感受出相关政策及管理条例等管理机制的颁布与实施,对药品注册申报数量有着直接的影响。而且,注册管理新政对药品注册分类进行改革和重新界定后,更利于制定针对性较强的文件进行管理与引导,相应的创新药品研发红利及优惠待遇也起到了积极促进的作用。如此一来,便能够使相关工作者与管理者能够更好地把握市场需求及临床需求,进而有效提升药品研发效率,避免相关资源的浪费,同时,也给企业带来一定的经济效益。
4.讨论及建议
我国药品注册管理新政实施后,对新药及仿制药的申报类别进行了科学的区分,同时对药品的研发热点也形成了一定的导向作用,还对药品注册申报资料内容与相关技术要求进行了调整和规定,对我国药品注册申报的数量产生了一定程度的影响[6]。
为了确保药品注册及申报的成功率和效率,药品研发企业应当对自身工作不断地改进和寻求转型机会,例如将新药的研发向临床需求及市场需求靠拢,同时改善药品研发的整体效率,规范相关内容资料,以便在进行评审审批的过程中,尽可能地节约药品审评申报的时间资源,提高药品注册申报的成功率,给企业带来经济效益。此外,还需注重在保障或提升药品质量的前提下,对药品进行适当创新和优化,多为人民群众着想,满足人民群众的用药需求,使其更符合市场需求。在进行药品研发的过程中,药品研发企业还应当加强对其过程中的监督与管理,即从立项阶段、研究阶段、临床研究阶段等,到最终的审批阶段与生产阶段,都需要严格遵循我国相关机制进行监管,按照国家的相关制度执行,确保药品的安全性和有效性。
结束语
药品注册的管理新政是促进医药行业稳步发展的重要举措,不断优化药品的研发申报相关流程,有效提升药品的研发质量,对药品质量和疗效方面有着极大的影响作用。因而,医药企业应当重视管理新政的作用,参照管理新政中的相关内容,立足于人们对药品的临床实际需求,不断对药品的品质进行提升以及对药品种类进行创新,进一步提升药品研发及其申报流程的合理性及完善性,提高药品研发注册申报的成功率,给企业带来经济效益的同时确保药品的安全性和有效性。
【参考文献】
[1]王爱迪.分析药品注册管理新政对药品研发申报的影响[J].中文科技期刊数据库(全文版)医药卫生,2021(7):0164-0166.
[2]药品注册管理办法.国家药品监督管理局,第27号,自2020年7月1日起施行.
[3]张蕴,金鑫.药品研发项目注册申报沟通管理研究[J].中国卫生产业,2020,17(32):62-64.
[4]陈燕霞,刘钊晖,蔡庆群,丘振文,杨忠奇.各地医疗机构中药制剂注册和备案申报要求浅析[J].中国医药导刊,2022,24(5):440-445.
[5]《2021年度药品审评报告》国家药品监督管理局 2022年6月1日发布.
[6]王奇巍,赖树清,郭文.药品注册管理新政对药品研发申报的影响分析[J].中国医药工业杂志,2017,48(11):1660-1665.

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2025 期刊网(www.qikanchina.com) 琼ICP备2021005105号



